![]()
Project flow#
LaminDB allows tracking data flow on the entire project level.
Here, we walk through exemplified app uploads, pipelines & notebooks following Schmidt et al., 2022.
A CRISPR screen reading out a phenotypic endpoint on T cells is paired with scRNA-seq to generate insights into IFN-γ production.
These insights get linked back to the original data through the steps taken in the project to provide context for interpretation & future decision making.

More specifically: Why should I care about data flow?
Data flow tracks data sources & transformations to trace biological insights, verify experimental outcomes, meet regulatory standards, increase the robustness of research and optimize the feedback loop of team-wide learning iterations.
While tracking data flow is easier when it’s governed by deterministic pipelines, it becomes hard when it’s governed by interactive human-driven analyses.
LaminDB interfaces workflow mangers for the former and embraces the latter.
Setup#
Init a test instance:
!lamin init --storage ./mydata
Show code cell output
✅ saved: User(id='DzTjkKse', handle='testuser1', email='testuser1@lamin.ai', name='Test User1', updated_at=2023-10-04 16:39:14)
✅ saved: Storage(id='LaHMxEPv', root='/home/runner/work/lamin-usecases/lamin-usecases/docs/mydata', type='local', updated_at=2023-10-04 16:39:14, created_by_id='DzTjkKse')
💡 loaded instance: testuser1/mydata
💡 did not register local instance on hub (if you want, call `lamin register`)
Import lamindb:
import lamindb as ln
from IPython.display import Image, display
💡 loaded instance: testuser1/mydata (lamindb 0.55.0)
Steps#
In the following, we walk through exemplified steps covering different types of transforms (Transform).
Note
The full notebooks are in this repository.
App upload of phenotypic data  #
#
Register data through app upload from wetlab by testuser1:
ln.setup.login("testuser1")
transform = ln.Transform(name="Upload GWS CRISPRa result", type="app")
ln.track(transform)
output_path = ln.dev.datasets.schmidt22_crispra_gws_IFNG(ln.settings.storage)
output_file = ln.File(output_path, description="Raw data of schmidt22 crispra GWS")
output_file.save()
Show code cell output
hello
💡 Transform(id='VUvyNlSP2gGxeu', name='Upload GWS CRISPRa result', type='app', updated_at=2023-10-04 16:39:17, created_by_id='DzTjkKse')
💡 Run(id='mBE6M42X84CpXBhpNGK6', run_at=2023-10-04 16:39:17, transform_id='VUvyNlSP2gGxeu', created_by_id='DzTjkKse')
hello
within hello
Hit identification in notebook  #
#
Access, transform & register data in drylab by testuser2:
ln.setup.login("testuser2")
transform = ln.Transform(name="GWS CRIPSRa analysis", type="notebook")
ln.track(transform)
# access
input_file = ln.File.filter(key="schmidt22-crispra-gws-IFNG.csv").one()
# identify hits
input_df = input_file.load().set_index("id")
output_df = input_df[input_df["pos|fdr"] < 0.01].copy()
# register hits in output file
ln.File(output_df, description="hits from schmidt22 crispra GWS").save()
Show code cell output
hello
hello
💡 Transform(id='DocWlf50vCGdSG', name='GWS CRIPSRa analysis', type='notebook', updated_at=2023-10-04 16:39:22, created_by_id='bKeW4T6E')
💡 Run(id='o8mBOGr1iWZQwZhLnHaQ', run_at=2023-10-04 16:39:22, transform_id='DocWlf50vCGdSG', created_by_id='bKeW4T6E')
hello
within hello
hello
hello
Inspect data flow:
file = ln.File.filter(description="hits from schmidt22 crispra GWS").one()
file.view_flow()
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
hello
hello
hello

Sequencer upload  #
#
Upload files from sequencer:
ln.setup.login("testuser1")
ln.track(ln.Transform(name="Chromium 10x upload", type="pipeline"))
# register output files of upload
upload_dir = ln.dev.datasets.dir_scrnaseq_cellranger(
"perturbseq", basedir=ln.settings.storage, output_only=False
)
ln.File(upload_dir.parent / "fastq/perturbseq_R1_001.fastq.gz").save()
ln.File(upload_dir.parent / "fastq/perturbseq_R2_001.fastq.gz").save()
ln.setup.login("testuser2")
Show code cell output
hello
💡 Transform(id='IaDMxS6Lwc5Hj8', name='Chromium 10x upload', type='pipeline', updated_at=2023-10-04 16:39:24, created_by_id='DzTjkKse')
💡 Run(id='HPT7LKlZ8UWMTkY3MrC3', run_at=2023-10-04 16:39:24, transform_id='IaDMxS6Lwc5Hj8', created_by_id='DzTjkKse')
hello
within hello
❗ file has more than one suffix (path.suffixes), inferring: '.fastq.gz'
❗ file has more than one suffix (path.suffixes), inferring: '.fastq.gz'
scRNA-seq bioinformatics pipeline #
Process uploaded files using a script or workflow manager: Pipelines and obtain 3 output files in a directory filtered_feature_bc_matrix/:
transform = ln.Transform(name="Cell Ranger", version="7.2.0", type="pipeline")
ln.track(transform)
# access uploaded files as inputs for the pipeline
input_files = ln.File.filter(key__startswith="fastq/perturbseq").all()
input_paths = [file.stage() for file in input_files]
# register output files
output_files = ln.File.from_dir("./mydata/perturbseq/filtered_feature_bc_matrix/")
ln.save(output_files)
Show code cell output
hello
💡 Transform(id='sH3y1OfXH46Qkq', name='Cell Ranger', version='7.2.0', type='pipeline', updated_at=2023-10-04 16:39:25, created_by_id='bKeW4T6E')
💡 Run(id='u1gUjLsnS2En4lqiZJEq', run_at=2023-10-04 16:39:25, transform_id='sH3y1OfXH46Qkq', created_by_id='bKeW4T6E')
hello
within hello
hello
hello
hello
hello
❗ file has more than one suffix (path.suffixes), inferring: '.tsv.gz'
❗ file has more than one suffix (path.suffixes), inferring: '.mtx.gz'
❗ file has more than one suffix (path.suffixes), inferring: '.tsv.gz'
Post-process these 3 files:
transform = ln.Transform(name="Postprocess Cell Ranger", version="2.0", type="pipeline")
ln.track(transform)
input_files = [f.stage() for f in output_files]
output_path = ln.dev.datasets.schmidt22_perturbseq(basedir=ln.settings.storage)
output_file = ln.File(output_path, description="perturbseq counts")
output_file.save()
Show code cell output
hello
❗ record with similar name exist! did you mean to load it?
| id | __ratio__ | |
|---|---|---|
| name | ||
| Cell Ranger | sH3y1OfXH46Qkq | 90.0 |
💡 Transform(id='xmujzZnXu2lvTS', name='Postprocess Cell Ranger', version='2.0', type='pipeline', updated_at=2023-10-04 16:39:25, created_by_id='bKeW4T6E')
💡 Run(id='cguYyM1vjsAHbuRnzJLP', run_at=2023-10-04 16:39:25, transform_id='xmujzZnXu2lvTS', created_by_id='bKeW4T6E')
hello
within hello
Inspect data flow:
output_files[0].view_flow()
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
hello
hello
hello
hello

Integrate scRNA-seq & phenotypic data #
Integrate data in a notebook:
transform = ln.Transform(
name="Perform single cell analysis, integrate with CRISPRa screen",
type="notebook",
)
ln.track(transform)
file_ps = ln.File.filter(description__icontains="perturbseq").one()
adata = file_ps.load()
file_hits = ln.File.filter(description="hits from schmidt22 crispra GWS").one()
screen_hits = file_hits.load()
import scanpy as sc
sc.tl.score_genes(adata, adata.var_names.intersection(screen_hits.index).tolist())
filesuffix = "_fig1_score-wgs-hits.png"
sc.pl.umap(adata, color="score", show=False, save=filesuffix)
filepath = f"figures/umap{filesuffix}"
file = ln.File(filepath, key=filepath)
file.save()
filesuffix = "fig2_score-wgs-hits-per-cluster.png"
sc.pl.matrixplot(
adata, groupby="cluster_name", var_names=["score"], show=False, save=filesuffix
)
filepath = f"figures/matrixplot_{filesuffix}"
file = ln.File(filepath, key=filepath)
file.save()
Show code cell output
hello
💡 Transform(id='AKgqswBfLxB9hi', name='Perform single cell analysis, integrate with CRISPRa screen', type='notebook', updated_at=2023-10-04 16:39:27, created_by_id='bKeW4T6E')
💡 Run(id='lPhlAwPuFLWp9U33s0Hr', run_at=2023-10-04 16:39:27, transform_id='AKgqswBfLxB9hi', created_by_id='bKeW4T6E')
hello
within hello
hello
hello
hello
hello
WARNING: saving figure to file figures/umap_fig1_score-wgs-hits.png
WARNING: saving figure to file figures/matrixplot_fig2_score-wgs-hits-per-cluster.png
Review results#
Let’s load one of the plots:
ln.track()
file = ln.File.filter(key__contains="figures/matrixplot").one()
file.stage()
Show code cell output
💡 notebook imports: ipython==8.16.1 lamindb==0.55.0 scanpy==1.9.5
💡 Transform(id='1LCd8kco9lZUz8', name='Project flow', short_name='project-flow', version='0', type=notebook, updated_at=2023-10-04 16:39:30, created_by_id='bKeW4T6E')
💡 Run(id='CHsRIb1j9VkCrXVmiY9J', run_at=2023-10-04 16:39:30, transform_id='1LCd8kco9lZUz8', created_by_id='bKeW4T6E')
hello
within hello
hello
hello
PosixUPath('/home/runner/work/lamin-usecases/lamin-usecases/docs/mydata/figures/matrixplot_fig2_score-wgs-hits-per-cluster.png')
display(Image(filename=file.path))
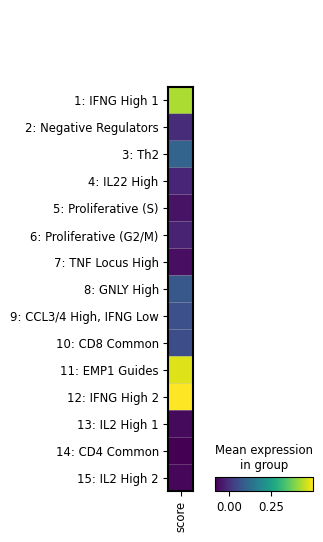
We see that the image file is tracked as an input of the current notebook. The input is highlighted, the notebook follows at the bottom:
file.view_flow()
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
within hello
hello
within hello
hello
within hello
hello
within hello
hello
hello
hello
hello
hello
hello
hello
hello
hello
hello
hello
hello
hello
hello
hello
hello
hello
hello

Alternatively, we can also look at the sequence of transforms:
transform = ln.Transform.search("Bird's eye view", return_queryset=True).first()
transform.parents.df()
hello
hello
within hello
| name | short_name | version | type | latest_report_id | source_file_id | reference | reference_type | initial_version_id | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||
| DocWlf50vCGdSG | GWS CRIPSRa analysis | None | None | notebook | None | None | None | None | None | 2023-10-04 16:39:22 | bKeW4T6E |
| xmujzZnXu2lvTS | Postprocess Cell Ranger | None | 2.0 | pipeline | None | None | None | None | None | 2023-10-04 16:39:25 | bKeW4T6E |
transform.view_parents()
hello
hello
hello
hello
hello
hello
hello
hello
hello
hello
hello

Understand runs#
We tracked pipeline and notebook runs through run_context, which stores a Transform and a Run record as a global context.
File objects are the inputs and outputs of runs.
What if I don’t want a global context?
Sometimes, we don’t want to create a global run context but manually pass a run when creating a file:
run = ln.Run(transform=transform)
ln.File(filepath, run=run)
When does a file appear as a run input?
When accessing a file via stage(), load() or backed(), two things happen:
The current run gets added to
file.input_ofThe transform of that file gets added as a parent of the current transform
You can then switch off auto-tracking of run inputs if you set ln.settings.track_run_inputs = False: Can I disable tracking run inputs?
You can also track run inputs on a case by case basis via is_run_input=True, e.g., here:
file.load(is_run_input=True)
Query by provenance#
We can query or search for the notebook that created the file:
transform = ln.Transform.search("GWS CRIPSRa analysis", return_queryset=True).first()
hello
And then find all the files created by that notebook:
ln.File.filter(transform=transform).df()
| storage_id | key | suffix | accessor | description | version | size | hash | hash_type | transform_id | run_id | initial_version_id | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||
| 8m3q6CtdkGbhkCoY4F6z | LaHMxEPv | None | .parquet | DataFrame | hits from schmidt22 crispra GWS | None | 18368 | TufBUAIQVzLPDJ4sCV_kTg | md5 | DocWlf50vCGdSG | o8mBOGr1iWZQwZhLnHaQ | None | 2023-10-04 16:39:22 | bKeW4T6E |
Which transform ingested a given file?
file = ln.File.filter().first()
file.transform
hello
Transform(id='VUvyNlSP2gGxeu', name='Upload GWS CRISPRa result', type='app', updated_at=2023-10-04 16:39:17, created_by_id='DzTjkKse')
And which user?
file.created_by
hello
User(id='DzTjkKse', handle='testuser1', email='testuser1@lamin.ai', name='Test User1', updated_at=2023-10-04 16:39:24)
Which transforms were created by a given user?
users = ln.User.lookup()
hello
ln.Transform.filter(created_by=users.testuser2).df()
| name | short_name | version | type | latest_report_id | source_file_id | reference | reference_type | initial_version_id | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||
| DocWlf50vCGdSG | GWS CRIPSRa analysis | None | None | notebook | None | None | None | None | None | 2023-10-04 16:39:22 | bKeW4T6E |
| sH3y1OfXH46Qkq | Cell Ranger | None | 7.2.0 | pipeline | None | None | None | None | None | 2023-10-04 16:39:25 | bKeW4T6E |
| xmujzZnXu2lvTS | Postprocess Cell Ranger | None | 2.0 | pipeline | None | None | None | None | None | 2023-10-04 16:39:25 | bKeW4T6E |
| AKgqswBfLxB9hi | Perform single cell analysis, integrate with C... | None | None | notebook | None | None | None | None | None | 2023-10-04 16:39:27 | bKeW4T6E |
| 1LCd8kco9lZUz8 | Project flow | project-flow | 0 | notebook | None | None | None | None | None | 2023-10-04 16:39:30 | bKeW4T6E |
Which notebooks were created by a given user?
ln.Transform.filter(created_by=users.testuser2, type="notebook").df()
| name | short_name | version | type | latest_report_id | source_file_id | reference | reference_type | initial_version_id | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||
| DocWlf50vCGdSG | GWS CRIPSRa analysis | None | None | notebook | None | None | None | None | None | 2023-10-04 16:39:22 | bKeW4T6E |
| AKgqswBfLxB9hi | Perform single cell analysis, integrate with C... | None | None | notebook | None | None | None | None | None | 2023-10-04 16:39:27 | bKeW4T6E |
| 1LCd8kco9lZUz8 | Project flow | project-flow | 0 | notebook | None | None | None | None | None | 2023-10-04 16:39:30 | bKeW4T6E |
We can also view all recent additions to the entire database:
ln.view()
Show code cell output
File
| storage_id | key | suffix | accessor | description | version | size | hash | hash_type | transform_id | run_id | initial_version_id | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| id | ||||||||||||||
| PDdTwykM7mr4lggF7byV | LaHMxEPv | figures/matrixplot_fig2_score-wgs-hits-per-clu... | .png | None | None | None | 28814 | H0Pxpa-fZOvigo74eXHZsQ | md5 | AKgqswBfLxB9hi | lPhlAwPuFLWp9U33s0Hr | None | 2023-10-04 16:39:29 | bKeW4T6E |
| IOLRy2s78M3NQ0ezVqRA | LaHMxEPv | figures/umap_fig1_score-wgs-hits.png | .png | None | None | None | 118999 | 1-WtAvRL1d_SSjZvMMOMkg | md5 | AKgqswBfLxB9hi | lPhlAwPuFLWp9U33s0Hr | None | 2023-10-04 16:39:29 | bKeW4T6E |
| uTdDb9R2RvGmJibwuv6V | LaHMxEPv | schmidt22_perturbseq.h5ad | .h5ad | AnnData | perturbseq counts | None | 20659936 | la7EvqEUMDlug9-rpw-udA | md5 | xmujzZnXu2lvTS | cguYyM1vjsAHbuRnzJLP | None | 2023-10-04 16:39:27 | bKeW4T6E |
| kwgnRHLbwok0FYnpAZ0f | LaHMxEPv | perturbseq/filtered_feature_bc_matrix/matrix.m... | .mtx.gz | None | None | None | 6 | PtjMi2heO_8hpvIga-slLw | md5 | sH3y1OfXH46Qkq | u1gUjLsnS2En4lqiZJEq | None | 2023-10-04 16:39:25 | bKeW4T6E |
| KHFw3UxNcey0DQjaFC9i | LaHMxEPv | perturbseq/filtered_feature_bc_matrix/barcodes... | .tsv.gz | None | None | None | 6 | 26C4BEGZStYCFyw2sdtejA | md5 | sH3y1OfXH46Qkq | u1gUjLsnS2En4lqiZJEq | None | 2023-10-04 16:39:25 | bKeW4T6E |
| 3t9knMSF3Hi8hCfcOvHZ | LaHMxEPv | perturbseq/filtered_feature_bc_matrix/features... | .tsv.gz | None | None | None | 6 | n-rZf_F77g-XKDGjfdfFfw | md5 | sH3y1OfXH46Qkq | u1gUjLsnS2En4lqiZJEq | None | 2023-10-04 16:39:25 | bKeW4T6E |
| GgLnkM74hYBmUqesNlM6 | LaHMxEPv | fastq/perturbseq_R2_001.fastq.gz | .fastq.gz | None | None | None | 6 | FvpUaB1m1DQ2cI7KABzjmQ | md5 | IaDMxS6Lwc5Hj8 | HPT7LKlZ8UWMTkY3MrC3 | None | 2023-10-04 16:39:24 | DzTjkKse |
Run
| transform_id | run_at | created_by_id | report_id | is_consecutive | reference | reference_type | |
|---|---|---|---|---|---|---|---|
| id | |||||||
| mBE6M42X84CpXBhpNGK6 | VUvyNlSP2gGxeu | 2023-10-04 16:39:17 | DzTjkKse | None | None | None | None |
| o8mBOGr1iWZQwZhLnHaQ | DocWlf50vCGdSG | 2023-10-04 16:39:22 | bKeW4T6E | None | None | None | None |
| HPT7LKlZ8UWMTkY3MrC3 | IaDMxS6Lwc5Hj8 | 2023-10-04 16:39:24 | DzTjkKse | None | None | None | None |
| u1gUjLsnS2En4lqiZJEq | sH3y1OfXH46Qkq | 2023-10-04 16:39:25 | bKeW4T6E | None | None | None | None |
| cguYyM1vjsAHbuRnzJLP | xmujzZnXu2lvTS | 2023-10-04 16:39:25 | bKeW4T6E | None | None | None | None |
| lPhlAwPuFLWp9U33s0Hr | AKgqswBfLxB9hi | 2023-10-04 16:39:27 | bKeW4T6E | None | None | None | None |
| CHsRIb1j9VkCrXVmiY9J | 1LCd8kco9lZUz8 | 2023-10-04 16:39:30 | bKeW4T6E | None | None | None | None |
Storage
| root | type | region | updated_at | created_by_id | |
|---|---|---|---|---|---|
| id | |||||
| LaHMxEPv | /home/runner/work/lamin-usecases/lamin-usecase... | local | None | 2023-10-04 16:39:14 | DzTjkKse |
Transform
| name | short_name | version | type | latest_report_id | source_file_id | reference | reference_type | initial_version_id | updated_at | created_by_id | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| id | |||||||||||
| 1LCd8kco9lZUz8 | Project flow | project-flow | 0 | notebook | None | None | None | None | None | 2023-10-04 16:39:30 | bKeW4T6E |
| AKgqswBfLxB9hi | Perform single cell analysis, integrate with C... | None | None | notebook | None | None | None | None | None | 2023-10-04 16:39:27 | bKeW4T6E |
| xmujzZnXu2lvTS | Postprocess Cell Ranger | None | 2.0 | pipeline | None | None | None | None | None | 2023-10-04 16:39:25 | bKeW4T6E |
| sH3y1OfXH46Qkq | Cell Ranger | None | 7.2.0 | pipeline | None | None | None | None | None | 2023-10-04 16:39:25 | bKeW4T6E |
| IaDMxS6Lwc5Hj8 | Chromium 10x upload | None | None | pipeline | None | None | None | None | None | 2023-10-04 16:39:24 | DzTjkKse |
| DocWlf50vCGdSG | GWS CRIPSRa analysis | None | None | notebook | None | None | None | None | None | 2023-10-04 16:39:22 | bKeW4T6E |
| VUvyNlSP2gGxeu | Upload GWS CRISPRa result | None | None | app | None | None | None | None | None | 2023-10-04 16:39:17 | DzTjkKse |
User
| handle | name | updated_at | ||
|---|---|---|---|---|
| id | ||||
| bKeW4T6E | testuser2 | testuser2@lamin.ai | Test User2 | 2023-10-04 16:39:25 |
| DzTjkKse | testuser1 | testuser1@lamin.ai | Test User1 | 2023-10-04 16:39:24 |
Show code cell content
!lamin login testuser1
!lamin delete --force mydata
!rm -r ./mydata
✅ logged in with email testuser1@lamin.ai and id DzTjkKse
💡 deleting instance testuser1/mydata
✅ deleted instance settings file: /home/runner/.lamin/instance--testuser1--mydata.env
✅ instance cache deleted
✅ deleted '.lndb' sqlite file
❗ consider manually deleting your stored data: /home/runner/work/lamin-usecases/lamin-usecases/docs/mydata